Drug vignettes: Remdesivir
August 10, 2020

Updated 23 September 2020, 20 October 2020
Robin E Ferner*†, Jeffrey K Aronson
On behalf of the Oxford COVID-19 Evidence Service Team
Centre for Evidence-Based Medicine, Nuffield Department of Primary Care Health Sciences,
University of Oxford
*University of Birmingham
†University College London
Correspondence to r.e.ferner@bham.ac.uk
Potential targets for antiviral drugs in COVID-19
RNA viruses replicate by binding to host cells, fusing with cell membranes, and entering cells. There they subvert the host’s cellular machinery to replicate, using the virus’s own RNA-dependent RNA polymerase (RdRp) [1].
Fusion of coronaviruses requires “docking” of specialized glycoproteins, “spikes”, which protrude from the viral surface and interact with specific molecules on the surface of host cells. The virus binds to a membrane bound enzyme, ACE-2, and is activated by a tranmsmembrane serine protease, TM; both of those are potential targets.
Spike proteins vary greatly from one type of coronavirus to another. In contrast, the RdRp is non-structural protein (nsp) 12 of the coronavirus genome (Figure 1), which is highly conserved among different strains, with 70–100% homology [2]. This means that the virus’s RdRp is a possible target for drugs intended to inhibit coronavirus replication.
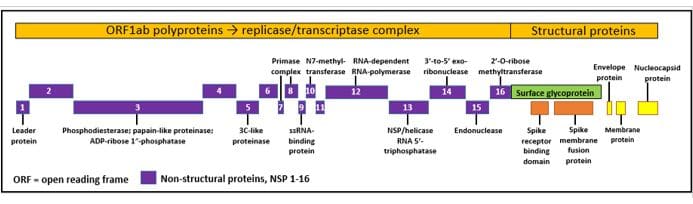
Figure 1. The genome of SARS-CoV-2
Possible therapeutic targets include the papain-like protease (PL pro, nsp 3), the 3C-like protease (3CL pro; nsp 5), the N7 methyltransferase (nsp 10), the RNA-dependent RNA polymerase (the RNA-dependent RNA polymerase (nsp 12), the helicase (nsp 13), and the receptor-binding domain (RBD)
One strategy, analogous to the use of cytosine arabinoside or 5-fluorouracil in cancer chemotherapy, is to develop nucleoside or nucleotide analogues whose intracellular phosphorylated derivatives inhibit viral nucleic acid synthesis. When the analogue is incorporated into a nucleic acid chain, it can lead to chain termination or to mismatch between the chain and its fellow, or in other ways interfere with the synthetic process [3,4]. A difficulty, however, is that coronaviruses possess an exoribonuclease (3′-5′ exonuclease, ExoN, nsp 14), a “proofreading” enzyme that corrects errors in the RNA sequence [5].
Remdesivir
A search for suitable analogues produced a long series of adenosine derivatives [6,7]. One adenosine-based compound, GS-441524, inhibits viral RdRp, but requires a rate-limiting phosphorylation step before it can enter cells. A phosphoramidate derivative, remdesivir (GS-5734) [8], by-passes this step, and allows ready entry into cells, where high concentrations of the active triphosphorylated derivative can accumulate (Figure 2) [9].
The pharmacokinetics of remdesivir in non-human primates indicates rapid uptake into cells [10]. The plasma half-life of the prodrug after intravenous injection is around 25 minutes and after 2 hours micromolar concentrations are achieved. The active drug has a half-life in monocytes of approximately 14 hours [11]. The half-maximal effective concentration for inhibition of viral replication in vitro is below 1 μM.

Figure 2. Activation of remdesivir
Remdesivir is a phosphoramidate analogue of adenosine; it enters the cell, where the phosphoramidate carrier is hydrolysed by esterases and the active triphosphorylated analogue of adenosine is formed by intracellular nucleoside-phosphate kinases
Remdesivir in infections other than SARS-CoV-2
Animal studies
Remdesivir inhibits MERS-CoV RdRp expressed in Spodoptera frugiperda cells, causing delayed RNA chain termination [12]. It is possible that the delay, which allows the addition of three or four further nucleotides to the RNA chain, protects the adenosine analogue from excision by ExoN. In the β-coronavirus murine hepatitis virus (MHV) model, the exonuclease provides the coronavirus some protection, as mutants that lack it are more susceptible to remdesivir, and mutations in the gene coding for ExoN can increase viral resistance to remdesivir [13]. However, the mutations so far encountered appear to confer a survival disadvantage on the virus.
Remdesivir has been tested in 18 rhesus monkeys infected with MERS-CoV [14]. Six animals were given vehicle only, six were treated with remdesivir 24 hours before inoculation, and six were treated 12 hours after inoculation. The control animals all developed respiratory signs, as did 5/6 animals treated after inoculation; but none of the animals given prophylactic doses developed respiratory signs. CT scans showed smaller and fewer lesions in the lungs after both prophylactic and therapeutic remdesivir.
Nipah virus, an RNA virus that causes fatal infection in humans, caused fatal infection in four African green monkeys given vehicle alone, but milder, non-fatal, disease in four animals given intravenous remdesivir 24 hours later [15].
Low-dose remdesivir (3 mg/kg) given intravenously for 12 days to rhesus monkeys reduced Ebola viraemia but did not uniformly protect them from fatal disease. A higher dose (10 mg/kg) did prevent fatal disease, even when given 3 days after infection [16].
Human studies
Human use of remdesivir in Ebola virus disease has been limited [17,18,19] The randomized controlled trial reported by Mulangu compared remdesivir with three different antibody treatments (REG-EB3, ZMapp, and Mab114) in 681 patients [20]. The mortality in the 175 patients treated with remdesivir was 53%, compared with 35% in the 174 patients treated with Mab114. The authors suggested that patients in the remdesivir group may have been ‘somewhat sicker’ at baseline.
Remdesivir in SARS-CoV-2
A “deep learning” algorithm examined potential treatments for SARS-CoV-2, and predicted that remdesivir would be active against several relevant viral enzymes, with a Kd for inhibition of the RdRp of 20 nM [21]. A molecular docking study predicted tight binding between remdesivir and the RdRp [22].
In cell culture, remdesivir inhibited viral replication, and a concentration of 0.77 μM produced half-maximal inhibition (EC50); the cytotoxic concentration was at least 100 times higher [23]. A review suggested an EC50 of about 23 μM [24].
Studies in patients
Holshue et al
The first patient to be treated for COVID-19 in the USA was a 35-year-old American who presented with cough and fever for four days, having returned from Wuhan on the first day of his illness. Viral PCR confirmed SARS-CoV-2 infection. The chest X-ray was clear 7 days after the start of the illness, but in view of fears of deterioration, remdesivir was infused. Radiology showed left lower lobe pneumonia on day 9, and a fall in oxygen saturation. He was given supplementary oxygen and vancomycin and cefepime for possible hospital-acquired pneumonia. His condition improved by day 12 and his symptoms gradually abated [25]. This case adds no useful information about the possible therapeutic value of remdesivir.
Bhatraju et al
The authors of an early report of 24 patients with confirmed COVID-19, of whom 18 required mechanical ventilation, stated that seven patients had received compassionate-use remdesivir, but noted that “we have insufficient information to report associated outcomes.” Twelve of the 24 patients died [26].
Grein et al
Short-term outcomes have been reported in 53 of 61 patients who received at least one dose of remdesivir, 200 mg on day 1 followed by 100 mg/day for the next nine days [27]. At the start of remdesivir treatment 30 patients were being ventilated, and four were having extracorporeal membrane oxygenation (ECMO). The patients were recruited from three continents. Follow-up was planned for day 28 or until discharge or death. The authors used a “cumulative index of clinical improvement”, but its validity is unclear. A more robust measure of outcome was death or discharge. By a median of 18 days after treatment had started, 25/53 patients had been discharged from hospital, and 7 (13%) had died. Among patients who received no mechanical ventilation, mortality was 5%. Sixty percent of patients suffered one or more adverse event, serious in 23%. The most common adverse events were abnormal liver function, diarrhoea, rash, renal impairment, and hypotension.
The authors noted that mortality in patients admitted to hospital in Wuhan was 22% overall and 66% in mechanically ventilated patients. As they stated, “Interpretation of the results of this study is limited by the small size of the cohort, the relatively short duration of follow-up, potential missing data owing to the nature of the program, the lack of information on 8 of the patients initially treated, and the lack of a randomized control group.”
Wang et al
This was a multi-site, randomized, masked, placebo-controlled trial of remdesivir + standard care versus standard care in patients with PCR positive SARS-CoV-2 infection, pneumonia on chest radiography, and hypoxyaemia (O2 saturation below 95% or a reduced PaO2:FiO2 ratio), recruited within 12 days of symptoms [28]. Recruitment stopped after 158 patients had been enrolled in the remdesivir arm and 78 in the placebo arm; a further patient, enrolled in the placebo arm, did not take part in the trial. The groups were somewhat mismatched, with more men in the placebo group (56% -v- 65%), but more patients with hypertension, diabetes, and coronary heart disease, and more patients who required high-flow oxygen or non-invasive ventilation in the remdesivir group. Rates of additional treatments—interferon alfa, lopinavir-ritonavir, antibacterial agents, and corticosteroids—were similar in the two groups. The primary end-point was defined as time to clinical improvement of two grades or more on a six-point scale, or discharge from hospital, within 28 days after randomization. The primary outcome was a non-significant difference in clinical improvement, which fell from 23 days in the placebo arm to 21 days in the remdesivir arm (hazard ratio 1.23, 0.87 to 1.75). Mortality by day 28 was 14% in the remdesivir arm and 13% in the placebo arm. The authors concluded that “Our trial found that intravenous remdesivir did not significantly improve the time to clinical improvement, mortality, or time to clearance of virus in patients with serious COVID-19 compared with placebo.” Note that the mortality rate was very similar to the rate reported by Grein et al in the case series of patients treated with remdesivir published in the New England Journal of Medicine [27].
Beigel et al
The preliminary results of the first stage of ACTT-1, an adaptive trial of treatments for COVID-19, were published on 22 May 2020 [29]. The results of the trial, in which 538 patients were randomized to remdesivir and 521 to placebo, were analysed early, at the request of the data monitoring committee. Financial support came from the National Institute of Allergy and Infectious Diseases, but Gilead Sciences provided remdesivir for the trial.
The specified primary outcome was time to recovery up to day 29, recovery having been defined in the protocol as “the first day on which the subject satisfies one of the following three categories from the ordinal scale: 1) Hospitalized, not requiring supplemental oxygen – no longer requires ongoing medical care; 2) Not hospitalized, limitation on activities and/or requiring home oxygen; 3) Not hospitalized, no limitations on activities.” Mortality at 28 days and duration of hospital stay were two of 28 secondary outcomes [30]. Selected baseline and outcome data are shown in Table 1.
Table 1. Selected data from the study of Beigel et al

Based on these results, the European Medicines Agency granted a Conditional Marketing Authorization to remdesivir [32].
The final report of this Gilead-sponsored trial was published on 15 October 2020.
The primary outcome was time to recovery up to day 29, based on the ordinal scale set out above. The initial report analysed only data up to day 15. The new report gives details of the 532 (of 541) patients allocated remdesivir and the 516 (of 521) patients allocated placebo, who died or were followed to day 29. 54 patients initially classified as having “mild-to-moderate” disease were reclassified to “severe” disease during the trial. Of those allocated remdesivir, 517 completed the study, and of those allocated placebo, 508 did so. There were some differences between the groups at baseline. In the placebo group, 26 patients received remdesivir; analyses evaluating the effects of unblinding and crossover produced results similar to the primary analysis.
A Kaplan-Meier analysis of cumulative recoveries by day 28 showed the following numbers still at risk:
|
Clinical Status |
Initial treatment |
Comparison |
| Remdesivir |
Placebo |
| Initial |
Final |
Initial |
Final |
Rate ratio |
95% CI |
| Overall |
541 |
84 |
521 |
105 |
1.29 |
1.10–1.53 |
| Receiving no oxygen |
75 |
2 |
63 |
2 |
1.29 |
0.91–1.83 |
| Receiving oxygen |
232 |
13 |
203 |
28 |
1.45 |
1.18–1/79 |
| NIVV or high-flow oxygen |
95 |
27 |
98 |
27 |
1-.09 |
0.76–1.57 |
| Ventilation or ECMO |
131 |
42 |
154 |
48 |
0.98 |
0.70–1.36 |
NIVV = non-invasive ventilation, ECMO = extracorporeal membrane oxygenation, CI = confidence interval
The recovery, as judged by change on the categorical scale used, was significantly better for the cohort overall, and for those patients who were ill enough to be given supplemental oxygen, but who did not require high-flow oxygen, ventilation, or ECMO. The group receiving supplemental oxygen made up 41% of the entire cohort.
The differences in mortality at day 29 were reported as follows:
|
Clinical Status |
Initial treatment |
Comparison |
| Remdesivir |
Placebo |
| Deaths, day 29 |
Deaths, day 29 |
Hazard ratio |
95% CI |
| Overall |
59 (11.4%) |
77 (15.2%) |
0.73 |
0.52–1.03 |
| Receiving no oxygen |
3 (4.1%) |
3 (4.8%) |
0.82 |
0.17–4.07 |
| Receiving oxygen |
9 (4.0%) |
25 (12.7%) |
0.30 |
0.14–0.64 |
| NIVV or high-flow oxygen |
19 (21.2%) |
20 (20.4%) |
1.02 |
0.54–1.91 |
| Ventilation or ECMO |
28 (21.9%) |
29 (19.3%) |
1.13 |
0.67–1.89 |
There was therefore no statistically significant difference in mortality at 15 days, as indicated in the first report of this study, or at 29 days. While there was a significant difference at the 5% level in mortality at 29 days for those receiving oxygen at the start of the trial, this was a subgroup analysis of a non-significant overall result, and no correction had been made for multiple tests.
The study confirmed an apparent benefit in time to recovery for patients treated with remdesivir of about 5 days. The supplementary tables show that 26 remdesivir-treated and 15 placebo-treated patients required re-admission, although a sensitivity analysis still showed the a statistically significant hazard ratio for recovery by 28 days, which was 1.22 (95% CI 1.05–1.41) in favour of remdesivir treatment.
Olender et al
The manuscript of a paper accepted for publication in the journal Clinical Infectious Diseases was posted online on 24 July 2020 [33]. The title was “Remdesivir for Severe COVID-19 versus a Cohort Receiving Standard of Care”. The corresponding author, who was not the first author, and a further 14 of the 33 named authors were from Gilead Sciences, the manufacturer of remdesivir, who provided funding for the study.
This was an analysis of observational data on the outcomes in patients with COVID-19, some of whom received treatment with remdesivir. The analysis included results from a study whose protocol was titled “Study to Evaluate the Safety and Antiviral Activity of Remdesivir (GS-5734™) in Participants With Severe Coronavirus Disease (COVID-19)” [34]. This was described as a randomized Phase III study. According to the clinicaltrials.gov website, it had recruited 4891 patients by the time the study closed on 30 June 2020.
The study reported in Clinical Infectious Diseases combined the results of a randomized trial of two different doses of remdesivir and a retrospective cohort study of clinical outcomes in patients receiving “standard of care”. All the patients had tested positive for SARS-CoV-2, had been admitted to hospital, and required oxygen for an oxygen saturation of 94% or less.
The primary endpoint was the proportion of patients who had recovered at 14 days, judged by rather complex criteria related to a 7-point clinical scale, on which “recovery was defined as having a score of 5–7 points for patients with a baseline score of 2–4, or a score of 6–7 for patients with a baseline score of 5, or a score of 7 for patients with a baseline score of 6”.
The authors adjusted for differences in baseline characteristics by “the inverse probability of treatment weighting procedure”, which involved propensity scores, and produced 312 patients (out of 397 patients assessed) treated with remdesivir and a control group of 818 patients (out of 1268 patients assessed) who received standard care. The number of remdesivir-treated subjects was 298 before statistical adjustment and 312 after adjustment. Notes stated that “The weighted patient number was 312 after applying the IPTW weighting method”, and “Based on IPTW, the number of patients in remdesivir and non-remdesivir cohorts were modestly different from the original sample size (some patients weighted more, and some patients weighted less based on the patients’ propensity scores)”.
Supplementary tables listed the factors for which correction was made. Supplemental Digital Content 6 was a table containing a list of potential medications for COVID-19 treatment; dexamethasone was not listed and was therefore presumably not included in the propensity scoring, despite the fact that it reduces mortality in those with severe disease.
A cohort of Italian patients was omitted because they had a higher mortality rate than expected.
The authors concluded that “In this comparative analysis, by day 14, remdesivir was associated with significantly greater recovery and 62% reduced odds of death versus standard-of-care treatment in patients with severe COVID-19.”
This is another company-sponsored interim analysis of observational data on remdesivir, when what we need is a proper, large, masked, randomized, controlled trial.
Spinner et al [added on 15 September 2020]
The results of an unmasked, three-arm, randomized trial of remdesivir for 10 days, remdesivir for 5 days, or standard care alone, were published online on 21 August 2020 [35] and in print on 15 September [36].
Patients were chosen to have “moderate COVID-19 pneumonia” and a positive PCR test for SARS-CoV-2. “Moderate COVID-19 pneumonia” was defined as the presence of any radiographic evidence of pulmonary infiltrates and an oxygen saturation above 94% on room air, with adequate liver and kidney function.
The treatments were randomly assigned in equal proportions to each of the three groups. The protocol specified “up to approximately 160 centers globally” and the number of subjects planned was “approximately 1600” [37]. However, in the published data 584 patients were described, recruited from 105 hospitals in the USA, Europe, and Asia; that is, fewer than six patients per hospital on average. A third of the hospitals enrolled 1 or 2 patients each.
Patients in the active treatment arms were given an intravenous infusion of remdesivir 200 mg on day 1 and 100 mg intravenously on subsequent days for 5 or 10 days.
The original primary objective (24 February 2020) was “To evaluate the efficacy of 2 remdesivir regimens compared to standard of care, with respect to the proportion of participants discharged on or before Day 14”; this was changed on 15 March to “To evaluate the efficacy of 2 remdesivir regimens compared to standard of care, with respect to clinical status assessed by a 7-point ordinal scale Day 11”.
The baseline data and primary and selected secondary outcome data were as follows:

OR = odds ratio, RR = rate ratio, NS = not significant at P=0.05
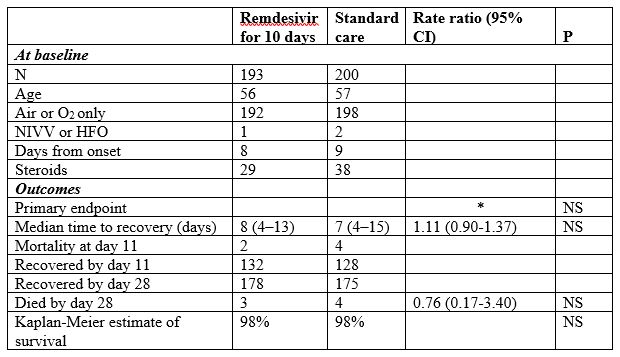
* “The proportional odds assumption was not met for the 10-day remdesivir group comparison, so no odds ratio is presented; the P value was calculated using the Wilcoxon rank sum test.”
“The 5-day or 10-day remdesivir groups and standard care did not differ significantly for time to clinical improvement, or time to recovery. The remdesivir and standard care groups did not differ significantly in duration of oxygen therapy or hospitalization, or in survival to 28 days.”
Eight of the thirty authors of the study were employees of Gilead, which markets remdesivir. A further nine authors had received financial or non-financial support from the company.
The authors interpreted their findings to mean that “Hospitalized patients with moderate COVID-19 randomized to a 5-day course of remdesivir had a statistically significantly better clinical status compared with those randomized to standard care at 11 days after initiation of treatment, but the difference was of uncertain clinical importance.”
The SOLIDARITY trial
The World Health Organization’s SOLIDARITY trial is a large prospective multicentre trial of remdesivir and other antiviral drugs in patients hospitalized with COVID-19. A pre-print of the results was published on 15 October 2020. Here we discuss the results for remdesivir against placebo.
Remdesivir was given in a dose of 200 mg on Day 0 and 100 mg/day on days 1–9. The primary endpoint was death at 28 days. Ventilation and time to discharge were also analysed.
There were 301 deaths in the 2743 patients allocated remdesivir and 303 deaths in the 2708 patients allocated placebo; rate ratio = 0.95 (95% CI 0.81–1.11, P = 0.50).
Ventilation was initiated in 295 of the remdesivir patients, and 284 of the placebo patients; 69% of the remdesivir cohort and 54% of the placebo cohort remained in hospital at day 7.
The authors also meta-analysed the effects of remdesivir, combining their own results with those of the ACCT-1, Wuhan, and SIMPLE trials. The risk difference in mortality between remdesivir and placebo was 0.7%, rate ratio 0.91 (95% CI 0.79–1.05).
They commented that “Although both ACTT-1 and Solidarity envisaged the possibility of different degrees of benefit in lower- and higher-risk patients, the particular lower-risk/higher-risk subdivision of the ACTT-1 findings in Figure 4 was unplanned. (The ACTT-1 protocol specified separate analyses of those not requiring any oxygen, with only 3/75 vs 3/63 deaths in ACTT-1, 11/661 vs 13/664 in Solidarity, and 5/384 vs 4/200 in SIMPLE; overall RR=0.82, but with wide confidence interval 0.43-1.55.) Thus, although the all-trials subtotals in Figure 4 suggest some benefit in low-risk patients and some hazard in high-risk inpatients (with the absolute benefit in low-risk appearing somewhat smaller than the absolute hazard in high-risk), neither subtotal should be considered in isolation from the other subtotal, or from the CI for the total.” We take this to mean that if the primary endpoint is not reached, then one should view with caution a post-hoc analysis that suggests that there is a subgroup of “responders” who showed significant benefit.
Meanwhile, the US National Institutes of Health (NIH) has issued a news article headed “Final report confirms remdesivir benefits for COVID-19”, with the key message that remdesivir shortened the time to recovery for hospital in-patients with COVID-19. It also stated, however, that “Remdesivir also improved mortality rates for those receiving supplemental oxygen (4% with remdesivir versus 13% with placebo at day 29 of treatment). All-cause mortality among all patients was 11% with remdesivir and 15% with placebo at day 29, but this difference between the treatment groups was not large enough to rule out chance. The preliminary findings hadn’t shown an effect on mortality.”
Remdesivir may shorten the duration of COVID-19 illness severe enough to require hospital treatment, but whether it reduces mortality is still unclear, despite trials involving six and a half thousand patients.
Registered clinical trials
When we first published this vignette on 10 August 2020, we were aware of 11 trials of remdesivir in COVID-19, of which one was single-masked and three double-masked. The number of patients to be studied in the masked trials was 2061 out of 22,437 in all (9.2%).
The current position
The results of the compassionate-use programme for remdesivir suggest, but do not prove, that the benefits of remdesivir may outweigh the harms it might cause. It is reminiscent of the early case reports of the success of penicillin in infective endocarditis [35]. However, the mortality rate from COVID-19 is, by the standards of acute infective endocarditis, relatively low, and so an open, uncontrolled trial will never be able to define the benefits or harms.
Does remdesivir save patients with COVID-19? A definitive answer to this question will only come from a sufficiently large randomized trial, or perhaps from a systematic review and meta-analysis of randomized trials, with mortality as a primary endpoint. Meanwhile, the suggestions of improved or more rapid recovery have led to use of remdesivir without secure information on its effectiveness or cost-effectiveness.
Disclaimer: This article has not been peer-reviewed; it should not replace individual clinical judgement and the sources cited should be checked. The views expressed in this commentary represent the views of the authors and not necessarily those of the host institution, the NHS, the NIHR, or the Department of Health and Social Care. The views are not a substitute for professional medical advice.